

Forschungsprojekte
CTLA4-Insuffizienz
In diesem Forschungsprojekt wird die Frage angegangen, warum manche CTLA4-Mutationsträger an einer Multi-Organ-Autoimmunerkrankung erkranken, während andere Familienmitglieder mit der gleichen Mutation nicht krank werden (reduzierte Penetranz der Erkrankung). Ziel dieses Projektes ist es die CTLA4-Defizienz molekular besser zu verstehen, um aus diesem Verständnis heraus eine optimale Therapie zu entwickeln. Insbesondere sollen genetische Faktoren identifiziert werden, die erklären, wie es bei dieser Erkrankung zu der reduzierten Penetranz und zu der variablen Expressivität des Erkrankungsbildes kommt. Hier stehen das HLA-System, der T-Zell-Rezeptor und Komponenten der T-Zell-Rezeptor Signaltransduktion im Mittelpunkt.
Die Erforschung der molekularen Mechanismen der CTLA4-Insuffizienz im Speziellen und von Multi-Organ-Autoimmunerkrankungen im Allgemeinen, sowie die Entdeckung neuer Gendefekte kann dafür genutzt werden, neuartige und zielgerichtete Therapien zu entwickeln, die auf jeden Patienten individuell abgestimmt werden können. Dadurch werden Forschung und Patientenversorgung besser miteinander verzahnt und die Lebensbedingungen der Patient*innen verbessert.


Immundysregulation aufgrund von Mutationen in NFKB1
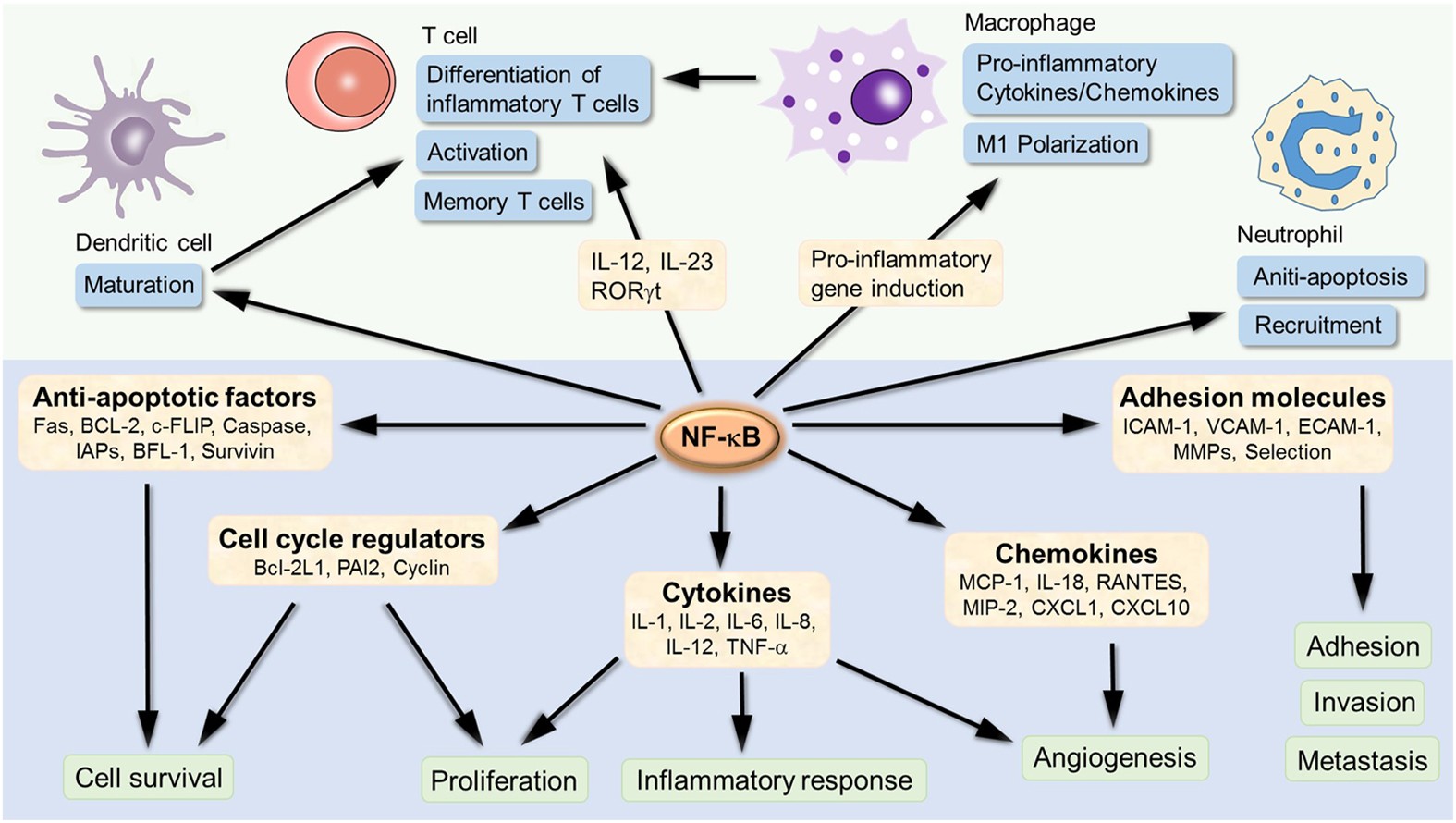
In diesem Forschungsprojekt sollen die klinische Erkrankung und die immunologischen Veränderungen von Patient*innen mit angeborenen Mutationen in NFKB1 beschrieben werden. NFkB1 ist ein Schlüsselmolekül in der Entwicklung, Regulation und Aktivierung des erworbenen und angeborenen Immunsystems (siehe Abbildung). Die feinabgestimmte Ko-expression und -regulation der verschiedenen Familienmitglieder der NFkB Familie ist von hoher Bedeutung für das Gleichgewicht des Immunsystems und der Entzündungsreaktion. Mutationen in NFKB1 sind die häufigste genetische Ursache des variablen Immundefektsyndroms (CVID). Es sollen neue diagnostische Untersuchungsverfahren der Erkrankung ermittelt werden, um die frühe und sichere Diagnose der Erkrankung und ihrer Aktivität besser nachzuweisen. Bei der Erkrankung spielen sekundäre entzündliche Signale wahrscheinlich eine wichtige Rolle. Die bessere Erkenntnis dieser Signale erlaubt die Planung gezielter Therapien in klinischen Studien, um so betroffenen Menschen langfristig eine personalisierte Therapie ihrer Erkrankung anbieten zu können.

Dr. Bärbel Keller
Universitätsklinikum Freiburg
Centrum für Chronische Immundefizienz (CCI)
Telefon: +49 (0)761 270-77691
E-Mail
Projekt 5: NFKB1

RELA
Dieses Forschungsprojekt zielt darauf ab, eine neue Typ 1-Interferonopathie zu charakterisieren, die durch heterozygote trunkierende RELA (p65)-Mutationen verursacht wird. Hierbei soll der Ausgangshypothese nachgegangen werden, dass die Interferenz des mutierten p65 mit NF-κB-Signalwegen die zelluläre Homöostase auf eine konstitutive Typ 1-IFN-Aktivierung ausrichtet. Daher werden die Auswirkungen von RELA-Mutationen auf pro- und antiinflammatorische Signalwege, die sich mit NF-κB und Typ 1-IFN-Signalwegen überschneiden, auf zellulärer, molekularer und transkriptioneller Ebene sowohl in Patientenzellen als auch in Gen-editierten Zellen untersucht. Gesamtziel des Projekts ist es, die zugrundeliegenden molekularen Mechanismen zu entschlüsseln und neue diagnostische und therapeutische Ziele zu identifizieren.


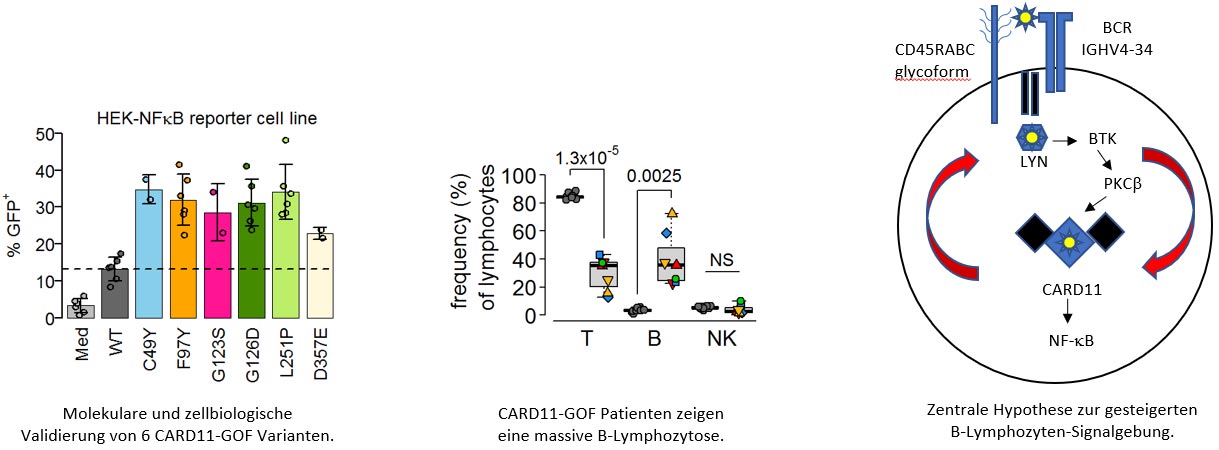
CARD11-GOF
In diesem Forschungsprojekt wird untersucht, welche genetischen, biochemischen und zellulären Veränderungen und Mechanismen der CARD11 gain-of-function Erkrankung zugrunde liegen. Insbesondere soll geklärt werden, warum es zu einer massiven Zunahme von B-Lymphozyten kommt, welche zur Multi-Organ-Autoimmunerkrankung führen. Es soll untersucht werden, ob bestimmte sich in der Entwicklung befindliche Substanzen künftig in die Therapie der CARD11 gain-of-function Erkrankung einfließen können.

Molekulare und klinische Konsequenzen von STAT3-aktivierenden Mutationen
Aktivierende Keimbahn-Mutationen in STAT3 verursachen Poly-Autoimmunität und Lymphoproliferation mit variabler Expressivität und inkompletter Penetranz. STAT3 überträgt Signale von Membranrezeptoren, insbesondere von Zytokinrezeptoren (z.B. IL-2, 6, 10, 11, 12, 15, 21, 23, 27) in den Zellkern. STAT3 regulierte Gene kontrollieren Zellproliferation, Überleben und Differenzierung von Immunzellen. Die Amplitude und Kinetik der STAT3 Signalgebung werden dabei von Phosphatasen und SOCS Proteinen moduliert.
In diesem Forschungsprojekt wird der Einfluss von verschiedenen Krankheits-relevanten aktivierenden STAT3 Mutationen auf die STAT3 Signalkaskade verglichen. Es wird Phosphorylierung und De-Phosphorylierung, Homo- und Hetero-Dimerisierung, nukleäre Translokation, DNA-Bindung und transkriptionelle Aktivierung untersucht. Zunächst soll herausgefunden werden, ob die spezifischen (variablen) molekularen Folgen der Mutationen einen Einfluss auf Expressivität und Penetranz der Krankheitserscheinungen haben. Danach sollen einige dieser gut charakterisierten Mutationen in primären T-Zellen überexprimiert werden und den Einfluss auf das biologische Verhalten der Zellen im Hinblick auf den klinischen Phänotyp beschreiben. Der Fokus liegt dabei insbesondere auf Proliferation und Zelltod. Ziel ist die Etablierung eines in vitro Systems, in dem eine gezielte pharmakologische Reversion der Überaktivierung des Signalwegs untersucht werden kann. Dadurch sollen neue Therapieansätze für Patient*innen mit aktivierenden STAT3 Mutationen gefunden werden.

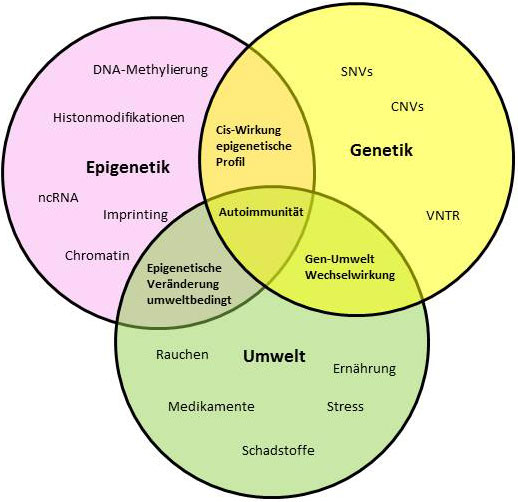
Identifizierung epigenetischer Faktoren bei Multi-Organ-Autoimmunerkrankungen
In den letzten Jahren haben Fortschritte in den Techniken der DNA-Sequenzierung zur Identifizierung einer wachsenden Anzahl an Genen geführt, die Immundefekte und Immundysregulation verursachen. Jedoch wirft die Identifizierung einer überraschend großen Anzahl von Mutationsträgern unter den klinisch gesunden Familienmitgliedern der Patient*innen Fragen auf, hinsichtlich modifizierender Faktoren, die möglicherweise für die reduzierte Penetranz der autosomal dominanten Erkrankungen wie NF-ĸB1-, CTLA4-Defizienz und STAT3-GOF verantwortlich sind. Die Tatsache, dass die meisten Multi-Organ-Autoimmunerkrankungen spät einsetzen, unterstreicht die Annahme, dass andere Faktoren wie Umwelteinflüsse, somatische Mutationen oder epigenetische Veränderungen eine Schlüsselrolle bei der Entstehung und dem Auftreten der Erkrankung spielen. Die epigenetischen Strukturen sind biochemische Bestandteile des Erbguts, die zusätzlich zur DNA wichtige Informationen für die Zelle enthalten.
In diesem Forschungsprojekt werden die möglichen epigenetischen Veränderungen untersucht, die erklären, wie es bei diesen Erkrankungen zu einer reduzierten Penetranz und variablem klinischem Erscheinungsbild kommt. Dies soll helfen die Patient*innen besser bei der Prognose ihrer Krankheit zu beraten und neuartige Strategien zur Vorbeugung der klinischen Manifestationen oder Komplikationen zu entwickeln.


Forschungsprojekte 2019 – 2022
Erstbeschreibung der humanen DGKZ-Defizienz
In diesem Forschungsprojekt wird untersucht, ob Veränderungen im Gen DGKZ eine Multi-Organ-Autoimmunerkrankung verursachen und über welche immunologischen Mechanismen diese vonstattengehen könnte. Es werden Patient*innen mit ursächlich ungeklärter Multi-Organ-Autoimmunerkrankung mit modernen genetischen Hochdurchsatztechniken auf bisher unbekannte genetische Veränderungen hin untersucht. Die so identifizierten genetischen Veränderungen werden mit zellbiologischen und molekularbiologischen Techniken analysiert, um Erkenntnisse zu grundlegenden Krankheitsmechanismen zu gewinnen. Basierend auf diesen Erkenntnissen werden immunmodulierende und immununterdrückende Medikamente auf ihre Wirksamkeit hin getestet, um die Grundlagen für präzise Behandlungen von Menschen mit Multi-Organ-Autoimmunerkrankungen zu schaffen.
Die Rolle von GARP als monogene Ursache von Multi-Organ-Autoimmunerkrankungen
Autoimmunerkrankungen entstehen, wenn es funktionell inkompetenten regulatorischen T-Zellen (Tregs) nicht gelingt, eine Expansion von aktivierten autoantigenspezifischen Effektor-T-Zellen zu verhindern.
Im diesem Forschungsprojekt wird eine detaillierte Analyse des Phänotyps und der molekularen Funktionen von Tregs bei Patienten mit Multi-Organ-Autoimmunerkrankungen erarbeitet, um mögliche „Achillesfersen“ für Treg-Funktionen zu identifizieren. In Vorarbeiten wurde eine Genchip-Analyse von Tregs durchgeführt und dabei konnte ein neues, Treg-spezifisches Oberflächenmolekül identifiziert werden, Glykoprotein A repetitions predominant (GARP). Eine eingeschränkte Expression von GARP ist mit einer beeinträchtigten Treg-Funktion assoziiert. Die Rolle von GARP bei monogenen Multi-Organ-Autoimmunerkrankungen ist derzeit nicht bekannt. Ein tiefgreifendes Verständnis dieser Rolle könnte dazu beitragen, unterschiedliche Mechanismen der Autoimmunität zu erkennen und damit die Tür zu einer besseren diagnostischen und therapeutischen Versorgung von Patient*innen zu öffnen. Die Funktionsanalyse von GARP zeigt eine Rolle als Rezeptor und Aktivator von latentem TGFß. Da TGFß für eine ordnungsgemäße Treg-Funktion entscheidend ist, wird im Detail die Rolle von GARP in Tregs mit einem Fokus auf TGFß-assoziierten Signalkaskaden untersucht. Da die GARP-Expression durch eine posttranskriptionelle Regulierung streng kontrolliert wird, ist es verlockend zu spekulieren, dass genetische Variationen in GARP zur Bildung von de novo miRNA Bindungsstellen führen könnten und damit die Entstehung von Multi-Organ-Autoimmunerkrankungen fördern würden. Es werden detaillierte Analysen von GARP bei Patient*innen mit Multi-Organ-Autoimmunerkrankungen durchgeführt. Zu diesem Zweck werden Sequenzierungsanalysen der kodierenden Regionen und der 3’UTR von GARP angefertigt.


Monogenetische Immun-dysregulations-Syndrome und ihr Einfluss auf das Plasmazell-Kompartiment
In diesem Forschungsprojekt beschäftigen wir uns mit den Auswirkungen von monogenetischen Immundefekten auf die Rolle und Funktion einer ganz bestimmten Gruppe von Immunzellen: B-Zellen und Plasmazellen bilden als Teil des „erworbenen Immunsystems“ das humorale, also Antikörper produzierende Immunkompartiment.
Plasmazellen können hierbei lernen, hochspezifische Antikörper gegen Krankheitserreger zu produzieren. Man unterscheidet zwei Gruppen. Die eine Gruppe ist die hochausgebildete schnelle Eingreiftruppe, die sehr gezielt bei einer Infektion reagieren kann. Die zweite Gruppe sorgt für langanhaltenden Schutz. Sie kann im Knochenmark Jahre überleben und ist dafür verantwortlich, dass bei bestimmten Impfungen auch nach vielen Jahren noch Antikörper im Blut nachweisbar sind.
Durch monogenetische Immundefekte kann die Fähigkeit des Körpers gestört sein, dieses Immungedächtnis auszubilden. Dabei handelt es sich um sehr komplexe Prozesse, bei denen neben B-Zellen und Plasmazellen auch viele andere Zelltypen beteiligt sind. Wenn einzelne Botenstoffe oder unterstützende Zellen fehlen – was bei einem Immundefekt möglich ist – kann das Immungedächtnis nicht so ausgebildet werden wie bei einem Gesunden. Ziel dieses Forschungsprojektes ist die Charakterisierung des Immunsystems bei monogenetischen Immundefekten wie CTLA4-Defizienz, LRBA-Defizienz, DGKZ-Defizienz oder Mutationen in den Genen STAT3, NFKB1 oder GARP.
Die Ergebnisse liefern nicht nur wichtige Erkenntnisse über mögliche Behandlungen der Erkrankungen, die zu neuen Therapieoptionen führen, sie können im Umkehrschluss auch Hinweise auf die physiologische Bedeutung der betroffenen Gene im Prozess der Generierung des Immungedächtnisses haben.

Prof. Dr. med. Bimba Franziska Hoyer
Universitätsklinikum Schleswig-Holstein, Campus Kiel
1. Medizinische Klinik , Abteilung für Rheumatologie und Klinische Immunologie und Exzellenzzentrum für Entzündungsmedizin
Telefon: +49 (0)431 500-22203
E-Mail
Website
Projekt 9: Plasmazellen
Projekt 11: Klinische Studie ABACHAI
